Results for “FDA” 465 found
The Adderall Shortage: DEA versus FDA in a Regulatory War
A record number of drugs are in shortage across the United States. In any particular case, it’s difficult to trace out the exact causes of the shortage but health care is the US’s most highly regulated, socialist industry and shortages are endemic under socialism so the pattern fits. The shortage of Adderall and other ADHD medications is a case in point. Adderall is a Schedule II controlled substance which means that in addition to the FDA and other health agencies the production of Adderall is also regulated, monitored and controlled by the U.S. Drug Enforcement Administration (DEA).
 The DEA aims to “combat criminal drug networks that bring harm, violence, overdoses, and poisonings to the United States.” Its homepage displays stories of record drug seizures, pictures of “most wanted” criminal fugitives, and heroic armed agents conducting drug raids. With this culture, do you think the DEA is the right agency to ensure that Americans are also well supplied with legally prescribed amphetamines?
The DEA aims to “combat criminal drug networks that bring harm, violence, overdoses, and poisonings to the United States.” Its homepage displays stories of record drug seizures, pictures of “most wanted” criminal fugitives, and heroic armed agents conducting drug raids. With this culture, do you think the DEA is the right agency to ensure that Americans are also well supplied with legally prescribed amphetamines?
Indeed, there is a large factory in the United States capable of producing 600 million doses of Adderall annually that has been shut down by the DEA for over a year because of trivial paperwork violations. The New York Magazine article on the DEA created shortage has to be read to be believed.
Inside Ascent’s 320,000-square-foot factory in Central Islip, a labyrinth of sterile white hallways connects 105 manufacturing rooms, some of them containing large, intricate machines capable of producing 400,000 tablets per hour. In one of these rooms, Ascent’s founder and CEO — Sudhakar Vidiyala, Meghana’s father — points to a hulking unit that he says is worth $1.5 million. It’s used to produce time-release Concerta tablets with three colored layers, each dispensing the drug’s active ingredient at a different point in the tablet’s journey through the body. “About 25 percent of the generic market would pass through this machine,” he says. “But we didn’t make a single pill in 2023.”
… the company has acknowledged that it committed infractions. For example, orders struck from 222s must be crossed out with a line and the word cancel written next to them. Investigators found two instances in which Ascent employees had drawn the line but failed to write the word.
 The causes of the DEA’s crackdown appears to be precisely the contradiction in its dueling missions. Ascent also produces opioids and the DEA crackdown was part of what it calls Operation Bottleneck, a series of raids on a variety of companies to demand that they account for every pill produced.
The causes of the DEA’s crackdown appears to be precisely the contradiction in its dueling missions. Ascent also produces opioids and the DEA crackdown was part of what it calls Operation Bottleneck, a series of raids on a variety of companies to demand that they account for every pill produced.
To be sure, the opioid epidemic is a problem but the big, multi-national plants are not responsible for fentanyl on the streets and even in the early years the opioid epidemic was a prescription problem (with some theft from pharmacies) not a factory theft problem (see figure at left). Maybe you think Adderall is overprescribed. Could be but the DEA is supposed to be enforcing laws not making drug policy. The one thing one can say for certain is that Operation Bottleneck has surely been a success in creating shortages of Adderall.
The DEA’s contradictory role in both combating the illegal drug trade and regulating the supply of legal, prescription drugs is highlighted by the fact that at the same as the DEA was raiding and shutting down Ascent, the FDA was pleading with them to increase production!
For Ascent, one of the more frustrating parts of being told by the government to stop making Adderall is that other parts of the government have pleaded with the company to make more. The company says that on multiple occasions, officials from the FDA asked it to increase production in response to the shortage, and that Ron Wyden, the Democratic senator from Oregon, also pressed Ascent for help. They received responses similar to those the company gave the stressed-out callers looking for pills: Ascent didn’t have any information. Instead, the company directed them to the DEA.
Surgery is Not FDA Regulated
What would happen if the FDA regulated pharmaceuticals much less than currently? I have pointed to one useful comparison, new uses of old drugs do not have to go through FDA required efficacy trials in the new use. In other words, new uses of old drugs are regulated for safety-only. Thus,
“off-label prescribing” provides a window on to what a world of safety-only FDA regulation would look like. Off-label prescribing surely results in errors and problems but overall physicians tell us that off-label prescribing is highly beneficial and critical to good medical care.
Maxwell Tabarrok points to another useful comparison, surgery. Surgery is not FDA regulated, despite having many of the same asymmetric information problems as pharmaceuticals. Some surgical procedures are surely ineffective and unsafe. Yet, once again, the FDA-absent surgery market appears beneficial overall and like other markets it improves over time with greater safety and more efficacy. For example,
In the US, the death rate from medical and surgical care complications declined by 39% from 1999 to 2009.
Would we be better off if every new surgical procedure had to go through FDA-required efficacy trials before it could be offered to consumers?
Neither of these comparison proves that a world with less FDA regulation would be a better world but both refute the stories of a world run amuck in the absence of the FDA. In essence, the reason is that the world contains many sources of approval, recommendation, certification and review beyond the FDA and these would grow in scope and stature absent the FDA.
See Maximum Progress for more.
Addendum: More excellent Tabarrok material: The Spice Must Flow: The Dutch-Portuguese War-Part 1.
Don’t Let the FDA Regulate Lab Tests!
I have been warning about the FDA’s power grab over lab developed tests. Lab developed tests have never been FDA regulated except briefly during the pandemic emergency when such regulation led to catastrophic consequences. Catastrophic consequences that had been predicted in advanced by Paul Clement and Lawrence Tribe. Despite this, for reasons I do not understand, the FDA plan is marching forward but many other people are starting to warn of dire consequences. Here, for example, is the executive summary from a letter by ARUP Laboratories, a non-profit enterprise of the University of Utah Department of Pathology:
ARUP urges the FDA to withdraw the proposed rule:
- The FDA proposal will reduce, an in many cases eliminate, access to safe and essential testing services, particularly for patients with rare diseases.
- Laboratory-developed tests are not devices as defined by the Medical Device Amendments of 1976, nor are clinical laboratories acting as manufacturers.
- The FDA does not have the statutory authority to regulate laboratory-developed tests.
- The FDA does not have the authority to regulate states, or state-owned entities. This is particularly relevant for the proposed rule regarding academic medical centers.
- The FDA’s regulatory impact analysis is flawed in its design, source information, methods. and conclusions, and it systematically overestimates purported benefits of the proposed rule and dramatically underestimates its cost to society, the healthcare industry, and the ability to provide ongoing essential laboratory services to patients.
- The proposed rule would significantly limit the ability of clinical laboratories to respond quickly to future pandemic, chemical, and/or radiologic public health threats.
- The proposed rule would not be easily implementable, and it would create an insurmountable backlog of submissions that would hinder diagnostic innovation.
- The proposed rule limits the practice of laboratory medicine.
- The FDA has not evaluated less restrictive, easily administered alternatives, such as CLIA reform. This is particularly relevant for common test modifications used in most hospital and academic medical center settings.”
Here is the American Hospital Association:
…we strongly believe that the FDA should not apply its device regulations to hospital and health system LDTs. These tests are not devices; rather, they are diagnostic tools developed and used in the context of patient care. As such, regulating them using the device regulatory framework would have an unquestionably negative impact on patients’ access to essential testing. It would also disrupt medical innovation in a field demonstrating tremendous benefits to patients and providers.
Here is Mass General Brigham, a non-profit hospital system, affiliated with Harvard, and the largest hospital-based research enterprise in the United States:
…we are concerned with the heavy regulatory burden of this proposal. In implementing any regulatory structure, policymakers must consider if the benefits outweigh the costs. Given that FDA predicts 50 percent of tests would require premarket review, and 5 percent will require premarket approval, we have serious concerns that the costs may outweigh the benefits. Given that many LDTs are hospital-based and will never be commercialized, hospitals will have little incentive or ability to develop future LDTs under this proposed rule as they will have little to no opportunity to offset the costs associated with these new regulatory requirements. We are concerned that the regulatory burden could have significant implications on responsible innovation especially for LDTs targeting rare conditions, or public health emergencies.
Two U.S. public lab directors personally reached out to me to ask me amplify the warning. Consider it amplified!
Today is the last day for public comment. Get your comments in!
AOC Gets on the Anti-FDA Bandwagon
At least when it comes to suncreen. As long-time readers will know, I have been complaining about FDA over-regulation of sunscreen for a decade! Maybe now that AOC is on the case things will change.
AOC’s sunscreen video is pretty good. One point she doesn’t stress is that requiring Americans to use more oily, less natural-feeling sunscreen can cause less use and thus more skin cancer. Even more important is the general issue of reciprocity or polycentric authority:
My rule is very simple. I don’t think the FDA is better than the EMA so if any drug or device is approved in Europe it ought to be available for purchase in the United States with a label saying “Approved by the EMA. Not approved by the FDA.” (By the way, we do have reciprocity type agreements with Canada and New Zealand for food so this would not be unprecedented.)
US sunscreens are far behind the rest of the world and our regulations aren’t necessarily making our sunscreens better or safer — but it doesn’t have to be this way! pic.twitter.com/vaZXpZ2a7S
— Rep. Alexandria Ocasio-Cortez (@RepAOC) August 11, 2023
The FDA Still Doesn’t Trust Women
The FDA has a long history of antipathy towards personal testing. The FDA has opposed personal pregnancy tests, HIV tests, genetic tests, and COVID tests, as I discussed in my article Testing Freedom. Well, the FDA is at it again:
NYTimes: At a hearing Tuesday to consider whether the Food and Drug Administration should authorize the country’s first over-the-counter birth control pill, a panel of independent medical experts advising the agency was left to reckon with two contradictory analyses of the medication called Opill.
During the eight-hour session, the manufacturer of the pill, HRA Pharma, which is owned by Perrigo, and representatives of many medical organizations and reproductive health specialists said that data strongly supported approval. They said that Opill, approved as a prescription drug 50 years ago, was safe, effective and easy for women of all ages to use appropriately — and that over-the-counter availability was sorely needed to lower the country’s high rate of unintended pregnancies.
In contrast, F.D.A. scientists questioned the reliability of company data that was intended to show that consumers would take the pill at roughly the same time every day and comply with directions to abstain from sex or temporarily use other birth control if they missed a dose. The agency seemed especially concerned about whether women with breast cancer or unexplained vaginal bleeding would correctly choose not to take Opill and whether adolescents and people with limited literacy would use it accurately.
Note carefully: The FDA isn’t worried that women won’t take the pill at the same time every day they are worried that women who get the pill without a prescription won’t take it at the same time every day. I guess in the FDA’s view women need some mansplaining to take birth control or at least some doctorplaining.
Dr. Westhoff suggested that for most women, there is no advantage to a doctor prescribing the pills because doctors don’t typically monitor patient adherence and often only see such patients once a year.
Similarly, I suspect that women with breast cancer will be concerned enough about their health to read the warning, Don’t Take This Pill if You Have Breast Cancer. Who knows, women with breast cancer might even ask their cancer physician or Google or their GP(T) about what foods and drugs to take and which to avoid.
If I didn’t know the FDA’s long history of opposing personal testing, I would think this simply bizarre but not trusting people with their own health decisions is practically in the FDA’s DNA.
The FDA’s Lab-Test Power Grab
The FDA is trying to gain authority over laboratory developed tests (LDTs). It’s a bad idea. Writing in the WSJ, Brian Harrison, who served as chief of staff at the U.S. Department of Health and Human Services, 2019-2021 and Bob Charrow, who served as HHS general counsel, 2018-2021, write:
We both were involved in preparing the federal Covid-19 public-health emergency declaration. When it was signed on Jan. 31, 2020, the intent was to cut red tape and maximize regulatory flexibility to allow a nimble response to an emerging pandemic.
Unknown to us, the next day the FDA went in the opposite direction: It issued a new requirement that labs stop testing for Covid-19 and first apply for FDA authorization. At that time, LDTs were the only Covid tests the U.S. had, and many were available and ready to be used in labs around the country. But since the process for emergency-use authorization was extremely burdensome and slow—and because, as we and others in department leadership learned, it couldn’t process applications quickly—many labs stopped trying to win authorization, and some pleaded for regulatory relief so they could test.
Through this new requirement the FDA effectively outlawed all Covid-19 testing for the first month of the pandemic when detection was most critical. One test got through—the one developed by the Centers for Disease Control and Prevention—but it proved to be one of the highest-profile testing failures in history because the entire nation was relying on the test to work as designed, and it didn’t.
When we became aware of the FDA’s action, one of us (Mr. Harrison) demanded an immediate review of the agency’s legal authority to regulate these tests, and the other (Mr. Charrow) conducted the review. Based on the assessment, a determination was made by department leadership that the FDA shouldn’t be regulating LDTs.
Congress has never expressly given the FDA authority to regulate the tests. Further, in 1992 the secretary of health and human services issued a regulation stating that these tests fell under the jurisdiction of the Centers for Medicare and Medicaid Services, not the FDA. Bureaucrats at the FDA have tried to ignore this rule even though the Supreme Court in Berkovitz v. U.S. (1988) specifically admonished the agency for ignoring federal regulations.
Loyal readers will recall that I covered this issue earlier in Clement and Tribe Predicted the FDA Catastrophe. Clement, the former US Solicitor General under George W. Bush and Tribe, a leading liberal constitutional lawyer, rejected the FDA claims of regulatory authority over laboratory developed tests on historical, statutory, and legal grounds but they also argued that letting the FDA regulate laboratory tests was a dangerous idea. In a remarkably prescient passage, Clement and Tribe (2015, p. 18) warned:
The FDA approval process is protracted and not designed for the rapid clearance of tests. Many clinical laboratories track world trends regarding infectious diseases ranging from SARS to H1N1 and Avian Influenza. In these fast-moving, life-or-death situations, awaiting the development of manufactured test kits and the completion of FDA’s clearance procedures could entail potentially catastrophic delays, with disastrous consequences for patient care.
Clement and Tribe nailed it. Catastrophic delays, with disastrous consequences for patient care is exactly what happened. Thus, Harrison and Charrow are correct, giving the FDA power over laboratory derived tests has had and will have significant costs.
FDA Deregulation Increases Safety and Innovation and Reduces Prices
In an important and impressive new paper, Parker Rogers looks at what happens when the FDA deregulates or “down-classifies” a medical device type from a more stringent to a less stringent category. He finds that deregulated device types show increases in entry, innovation, as measured by patents and patent quality, and decreases in prices. Safety is either negligibly affected or, in the case of products that come under potential litigation, increased.
After moving from Class III (high regulation) to II (moderate), device types exhibited a 200% increase in patenting and FDA submission rates relative to control groups. Patents filed after these events were also of significantly higher quality, as measured by a 200% increase in received citations and market valuations. These effects do not spill over into similar device types.1 For Class II to I deregulations, the rate of patent filings increased by 50%, though insignificantly, and the quality of patent filings exhibited a significant 10-fold improvement, suggesting that litigation better promotes innovation.
…Down-classification yields considerable benefits, as the proponents of deregulation would predict, but what of product safety? Perhaps counterintuitively, I find that deregulation can improve product safety by exposing firms to more litigation. Despite some adverse event rates increasing after Class III to II events (albeit insignificantly), Class II to I events are associated with significantly lower adverse event rates.3 My analysis of patent texts also reveals that inventors focus more on product safety after deregulation. These results suggest that litigation encourages product safety more than regulation…
Some background. Medical devices are regulated under three categories. New types of devices (new, not necessarily high risk) are highly regulated Class III devices which must go through a pre-market approval process to prove safety and efficacy (like new drugs). The pre-market approval process is time-consuming and expensive but it comes with one significant benefit, federal preemption of state tort action, i.e. these devices are shielded from product liability. Class II devices are devices that are judged to be substantially equivalent to an already approved device–proving equivalence also takes time and money but it’s less onerous than proving safety and efficacy de novo. Note that device manufacturers often make their devices less innovative so they can be approved as Class II devices rather than as Class III devices. Class II devices are mostly also protected against tort litigation. Class I devices are not FDA-approved and are subject to tort litigation.
As experience develops with new devices such that new devices turn out to be not especially risky, the FDA sometimes deregulates or down-classifies these devices from Class III to Class II or from Class II to Class I. Rogers studies these down-classifications by comparing what happens to the down-classified device category to a control group of similar devices that were not down-classified. The control group is critical and Rogers shows that his results are robust to defining the control group in a variety of plausible ways. Some of the key results are shown in the following figure:
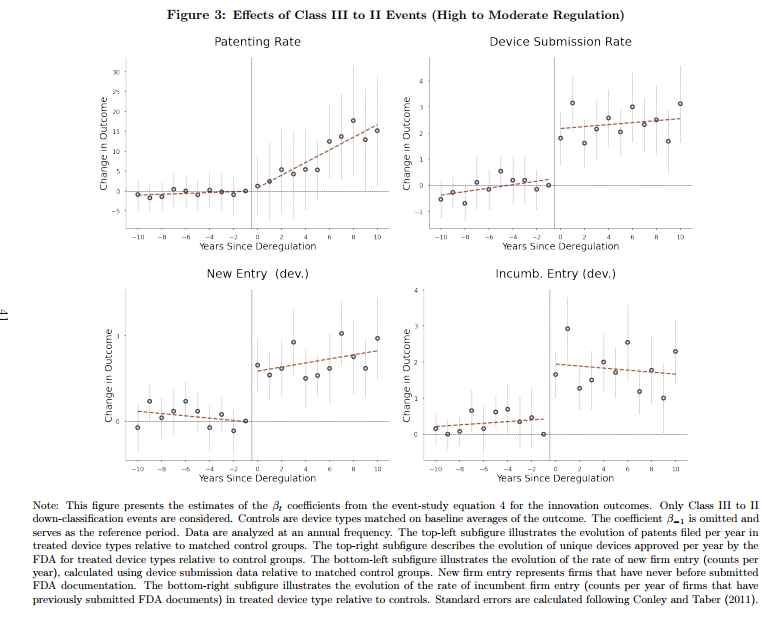 Nicely we see that device submissions and new entry occur very quickly once a device is down-regulated which indicates that firms have ideas and products on-the-shelf but they are dissuaded from entering the market by the onerous pre-market approval process. Most likely, these are products and firms which produce devices for the European market which tends to be less regulated and they enter the US market only when costs are reduced. Patenting also increases in the down-regulated device category and–exactly as one would expect–this takes more time.
Nicely we see that device submissions and new entry occur very quickly once a device is down-regulated which indicates that firms have ideas and products on-the-shelf but they are dissuaded from entering the market by the onerous pre-market approval process. Most likely, these are products and firms which produce devices for the European market which tends to be less regulated and they enter the US market only when costs are reduced. Patenting also increases in the down-regulated device category and–exactly as one would expect–this takes more time.
Safety declines non-significantly if at all from Class III to Class II deregulations and increases for Class II to Class I deregulations. That makes the welfare comparisons easy because deregulation appears to be all benefit and no cost. Note, however, that I have always argued that drugs and devices are actually too safe–that, is we could save more lives on net by approving more drugs and devices even if safety went down. That’s a hard sell, however, but it’s clearly true that given the results here we should deregulate or down-classify many more products even if safety declined on the margin. Too much safety is risky. That’s also the upshot of my paper on off-label prescribing which shows that it’s often the FDA-unapproved off-label use which is the gold-standard treatment in fast moving fields of medicine.
Rogers argues that safety increases for Class II to Class I deregulations because liability is a stronger deterrent on the margin than regulation (and he provides some evidence for this view in that safety increases more among larger firms that are less judgment proof than small firms). Without denying that mechanism my view is that innovation itself increases safety. As I noted above, medical device manufactures often do not use the latest technology in their products because this would threaten the “substantial equivalence” test so you get devices that are actually less safe and also more costly to manufacture than necessary. In essence, substantial equivalence anchors new technologies to old technologies thus preventing movement, even movement towards safety and lower prices.
Rogers also has an excellent and unusual paper (with Jeffrey Clemens) on directed innovation in artificial limbs due to the civil war! That paper and this one show a real focus on digging deep into the data to unearth important and unusual sources of insight. N.B.! Parker Rogers is on the job market.
Addendum: See my many previous posts for more useful references on the FDA, especially Is the FDA Too Conservative or Too Aggressive.
A Pox on the FDA
Monkeypox isn’t in the same category of risk that COVID was before vaccines but it’s a significant risk, especially in some populations, and it’s a test of how much we have learned. The answer is not bloody much. Here’s James Walsh in NYMag:
As monkeypox cases have ticked up nationwide, the White House and federal agencies have repeatedly assured the public that millions of vaccine doses will be distributed to at-risk populations before the end of the year. Yet since the World Health Organization announced the global monkeypox outbreak in May, only tens of thousands of shots have been administered in the U.S. The slow start is due, at least in part, to the fact that 1.1 million doses have been stored in a Denmark pharmaceutical facility while the Food and Drug Administration has taken almost two months to approve their release here, according to people familiar with the situation. FDA officials only began to inspect the facility last week. The lag time, public-health experts say, is indicative of the federal government’s lackadaisical approach to a growing public-health emergency.
…It’s unclear why the FDA took so long to send inspectors to Denmark. The agency regularly conducted virtual inspections of drug facilities early in the COVID-19 pandemic, according to the agency’s guidance, and public-health activists are demanding answers. “Members of at risk communities are being turned away from monkeypox vaccination because these vaccines are not available in sufficient quantity in the U.S., but instead sitting in freezers in Denmark,” members of the advocacy group PrEP4All and Partners in Health wrote in a letter to federal officials overseeing the outbreak response last week.
Compounding their frustrations was the FDA’s refusal to accept an inspection done last year by its counterpart, the European Medicines Agency, which deemed the company’s facility in compliance with the FDA’s own standards.
“The FDA does not grant reciprocity for EMA authorization of any vaccines, for monkeypox or other diseases,” a spokesperson for the FDA said in a statement.
Is there anyone in the United States who is saying, “I am at risk of Monkeypox and I want the vaccine but I don’t trust the European Medicines Agency to run the inspection. I’d rather wait for the FDA!” I don’t think so. James Krellenstein, an activist on this issue, asks:
“Why were the Europeans able to inspect this plant a year ago, ensuring these doses can be used in Europe and the Biden Administration didn’t do the same,” he added. “The FDA is making a judgment that they’d rather let gay people remain unvaccinated for weeks and weeks and weeks than trust the European certification process.”
Many people want to be vaccinated:
New York City has received just 7,000 doses from the federal government amid the national vaccine shortage. Meanwhile, the city Department of Health and Mental Hygiene’s appointment booking system has failed to keep up with the high demand for the shots — most recently on Wednesday.
…The mounting frustrations left health officials and Mayor Eric Adams on the defensive, pushing back against comparisons to New York’s struggles during the early days of the coronavirus vaccine, which was beset by computer glitches and supply shortages.
This is a classic case for reciprocity. Any drug, vaccine, test or sunscreen (!) approved by a stringent regulatory authority ought to be conditionally approved in the United States.
Addendum: If you are not furious already–and you should be–remember that during COVID the FDA suspended factory inspections around the world creating shortages of life-saving cancer drugs and other pharmaceuticals. As I wrote then “Grocery store workers are working, meat packers are working, hell, bars and restaurants are open in many parts of the country but FDA inspectors aren’t inspecting. It boggles the mind.”
Hat tip: Josh Barro.
Photo Credit: Nigeria Centre for Disease Control.
The FDA is Increasing Skin Cancer
Americans who travel to the beaches in France, Spain, or Italy routinely do something that is illegal in the United States–they buy and use European sunscreens to protect themselves from sunburn and skin cancer. Suncreens in Europe and Asia are better than in the United States because more ingredients are allowed and these create more effective and more pleasing suncreens. I’ve been writing about this since 2013! My view hasn’t changed:
My rule is very simple. I don’t think the FDA is better than the EMA so if any drug or device is approved in Europe it ought to be available for purchase in the United States with a label saying “Approved by the EMA. Not approved by the FDA.” (By the way, we do have reciprocity type agreements with Canada and New Zealand for food so this would not be unprecedented.)
Here’s the latest from Amanda Mull writing in the Atlantic:
Newer, better UV-blocking agents have been in use in other countries for years. Why can’t we have them here?
…In formal statements and position papers, doctors and cancer-prevention advocates express considerable interest in bringing new sunscreen ingredients to the American market, but not a lot of optimism that any will be available soon.
…In 2014, Congress passed a law attempting to speed access to sunscreen ingredients that have been in wide use in other countries for years, but it hasn’t really worked. “The FDA was supposed to be fast-tracking these ingredients for approval, because we have the safety data and safe history of usage from the European Union,” Dobos said. “But it seems to continually be stalled.” According to Courtney Rhodes, a spokesperson for the FDA, manufacturers have submitted eight new active ingredients for consideration. The agency has asked them to provide additional data in support of those applications, but none of them has yet satisfied the agency’s requirements.
“In the medical community, there is a significant frustration about the lack of availability of some of the sunscreen active ingredients,” Henry Lim, a dermatologist at Henry Ford Health, in Michigan, told me. The more filters are available to formulators, the more they can be mixed and matched in new ways, which stands to improve not just the efficacy of the final product, but how it feels and looks on your skin, and how easy it is to apply. On a very real level, making sunscreen less onerous to use can make it more effective. “The best sunscreen is going to be the one you’re going to use often and according to the directions,” Dobos said. Skin cancer is the most common type of cancer in the United States, and by one estimate, one in five Americans will develop it in their lifetime.
Hat tip: Joe.
The FDA should make Paxlovid easier to get
Pharmacists still cannot prescribe the medication themselves, a step that would cut the time it takes patients to secure the drug.
The Food and Drug Administration “is looking at this and thinking about it,” Dr. Jha said. “Whether they’re going to make a change, when and how, etc., is totally in their wheelhouse.”
Many patients are still handling the sometimes-cumbersome steps on their own: locating a virus test, then securing a Paxlovid prescription from a health provider, then finding a pharmacy that carries the pill, all within days of first showing symptoms.
Dr. Jha described being frustrated by physician colleagues who have told him they still limit Paxlovid to patients 65 years and older.
But no they still will not do this. I repeat myself, but you need to keep in mind the only time panel members have resigned from the FDA is when the Biden administration pushed through the booster shots.
Here is the full NYT article, via Rich Berger.
ProPublica on the FDA and Rapid Tests
Lydia DePillis has written the best piece on the FDA that I have ever read in a mainstream news publication. It gets everything right and yes it frankly verifies everything that I have been saying about the FDA and rapid tests for the last year and a half. I wish it had been written earlier but I suppose that illustrates how difficult it is to radically change people’s mindset from the FDA as protector to the FDA as threat. The sub head is:
Irene Bosch developed a quick, inexpensive COVID-19 test in early 2020. The Harvard-trained scientist already had a factory set up. But she was stymied by an FDA process experts say made no sense.
The piece recounts how cheap, rapid tests could have been approved in March of 2020! Here’s the opening bit:
When COVID-19 started sweeping across America in the spring of 2020, Irene Bosch knew she was in a unique position to help.
The Harvard-trained scientist had just developed quick, inexpensive tests for several tropical diseases, and her method could be adapted for the novel coronavirus. So Bosch and the company she had co-founded two years earlier seemed well-suited to address an enormous testing shortage.
E25Bio — named after the massive red brick building at MIT that houses the lab where Bosch worked — already had support from the National Institutes of Health, along with a consortium of investors led by MIT.
Within a few weeks, Bosch and her colleagues had a test that would detect coronavirus in 15 minutes and produce a red line on a little chemical strip. The factory where they were planning to make tests for dengue fever could quickly retool to produce at least 100,000 COVID-19 tests per week, she said, priced at less than $10 apiece, or cheaper at a higher scale.
“We are excited about what E25Bio is capable of shipping in a short amount of time: a test that is significantly cheaper, more affordable, and available at-home,” said firm founder Vinod Khosla. (Disclosure: Khosla’s daughter Anu Khosla is on ProPublica’s board.)
On March 21 — when the U.S. had recorded only a few hundred COVID-19 deaths — Bosch submitted the test for emergency authorization, a process the Food and Drug Administration uses to expedite tests and treatments.
You know how the story ends but really READ the WHOLE THING.
ProPublica on FDA Delay
If you have been following MR for the last 18 months (or 18 years!) you won’t find much new in this ProPublica piece on FDA delay in approving rapid tests but, other than being late to the game, it’s a good piece. Two points are worth emphasizing. First, some of the problem has been simple bureaucratic delay and inefficiency.
![]()
In late May, WHPM head of international sales Chris Patterson said, the company got a confusing email from its FDA reviewer asking for information that had in fact already been provided. WHPM responded within two days. Months passed. In September, after a bit more back and forth, the FDA wrote to say it had identified other deficiencies, and wouldn’t review the rest of the application. Even if WHPM fixed the issues, the application would be “deprioritized,” or moved to the back of the line.
“We spent our own million dollars developing this thing, at their encouragement, and then they just treat you like a criminal,” said Patterson. Meanwhile, the WHPM rapid test has been approved in Mexico and the European Union, where the company has received large orders.
An FDA scientist who vetted COVID-19 test applications told ProPublica he became so frustrated by delays that he quit the agency earlier this year. “They’re neither denying the bad ones or approving the good ones,” he said, asking to remain anonymous because his current work requires dealing with the agency.
Recall my review of Joseph Gulfo’s Innovation Breakdown.
Second, the FDA has engaged in regulatory nationalism–refusing to look at trial data from patients in other countries. This is madness when India does it and madness when the US does it.
For example, the biopharmaceutical giant Roche told ProPublica that it submitted a home test in early 2021, but it was rejected by the FDA because the trials had been done partly in Europe. The test had compared favorably with Abbott’s rapid test, and received European Union approval in June. The company plans to resubmit an application by the end of the year.
A smaller company, which didn’t want to be named because it has other contracts with the U.S. government, withdrew its pre-application for a rapid antigen test with integrated smartphone-based reporting because it heard its trial data from India — collected as the delta variant was surging there — wouldn’t be accepted. Doing the trials in the U.S. would have cost millions.
Photo credit: MaxPixel.
Clement and Tribe Predicted the FDA Catastrophe

Laboratory developed tests are not FDA regulated–never have been–instead the labs are regulated under the Clinical Laboratory Improvement Amendments (CLIA) as overseen by the CMS. Laboratory developed tests are the kind your doctor orders, they are a service not a product and are not sold directly to patients. Labs develop new tests routinely and they do not apply to the FDA for approval. Despite this long history, the FDA has claimed that it has the right to regulate lab tests and they have merely chosen not to exercise this right for forty years. In 2015, Paul Clement, the former US Solicitor General under George W. Bush, and Laurence Tribe, considered by many to be the leading constitutional lawyer in the United States, wrote an article that rejected the FDA’s claims writing that the “FDA’s assertion of authority over laboratory-developed testing services is clearly foreclosed by the FDA’s own authorizing statute” and “by the broader statutory context.”
Despite lacking statutory authority, the FDA has continued to claim it is authorized to regulate laboratory tests. Indeed, a key failure in the pandemic happened when the FDA issued so-called “guidance documents” saying that any SARS-CoV-II test had to be pre-approved by the FDA. Thus, the FDA reversed the logic of emergency. In ordinary times, pre-approval was not necessary but when speed was of the essence it became necessary to get FDA pre-approval. The FDA’s pre-approval process slowed down testing in the United States and it wasn’t until after the FDA lifted its restrictions in March that tests from the big labs became available.
Clement and Tribe rejected the FDA claims of regulatory authority over laboratory developed tests on historical, statutory, and legal grounds but they also argued that letting the FDA regulate laboratory tests was a dangerous idea. In a remarkably prescient passage, Clement and Tribe (2015, p. 18) warned:
The FDA approval process is protracted and not designed for the rapid clearance of tests. Many clinical laboratories track world trends regarding infectious diseases ranging from SARS to H1N1 and Avian Influenza. In these fast-moving, life-or-death situations, awaiting the development of manufactured test kits and the completion of FDA’s clearance procedures could entail potentially catastrophic delays, with disastrous consequences for patient care.
Clement and Tribe nailed it. Catastrophic delays, with disastrous consequences for patient care is exactly what happened.
Addendum: See also my pre-pandemic piece on this issue, Our DNA, Our Selves.
FDA relents on mix and match for third dose
Here is the NYT account, they sound both confused and confusing. How about “if you have had J&J, it is fine and probably preferable to get a further dose of Moderna or Pfizer”? Yet suddenly it is fine.
And it is the usual story — people have been doing this for months, and the FDA would not say it is terrible. Because they knew it wasn’t. But they wouldn’t say so. And now the status quo has shifted, and so everyone will treat it as fine, as if the supposed fears of yesterday never ever existed.
Maybe I should insult people more often?
FDA Approves American Rapid Antigen Test
What makes the FDAs failure to approve more rapid antigen tests even more galling is that the test being sold cheaply in the Amsterdam supermarket is the Flowflex, an American test made by Acon Labs in San Diego.
Well the FDA has finally approved the Acon test! Apparently it is good enough for the Germans and for US citizens. Hoorah! USA Today notes:
ACON expects to make 100 million tests per month by the end of this year. Production could double to 200 million monthly tests by February, according to the FDA.
…The United Kingdom and Germany have made significant purchases of home tests and widely distributed them to their residents to slow the spread of coronavirus. Such large government purchases allowed manufacturers to continue making tests even when demand softened as cases dropped.
The Biden administration will spend nearly $1.2 billion to purchase up to 187 million home tests from Abbott Laboratories and Celltrion Inc., company officials confirmed. The Department of Defense announced additional contracts totaling $647 million to buy 60 million kits from Abbott and three other testing vendors: OraSure Technologies, Quidel and Intrivio Holdings.
The FDA has authorized seven antigen-based tests that can be used at home without a prescription. The EU has authorized 21 tests beginning with the letter A (I am not sure all of these are authorized for home use but you get the idea.) Turtle slow. Still this is a big improvement.
Frankly, I think all the pressure from people like Michael Mina amplified by myself and others over 18 months and culminating in David Leonhartd’s NYTimes article Where Are the Tests? finally pushed them over the edge.