Rating the FDA by Division: Comparison with EMA
Reporters have been asking the FDA about my paper with DiMasi and Milne, AN FDA REPORT CARD. As you may recall, the upshot of that paper is that there is wide variance in the performance of FDA divisions. Here, for example, is the mean time to approval across divisions. 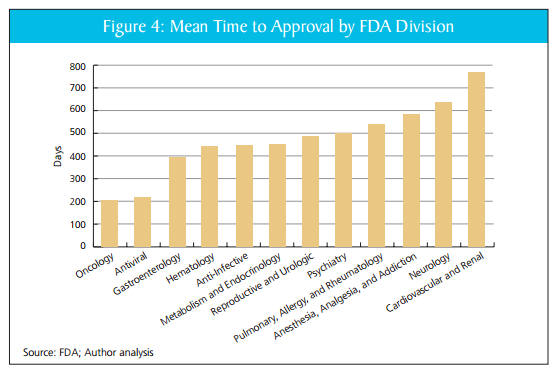
Our simple index, discussed in the paper, suggests that these differences are not easily explained by factors such as resources, complexity of task or differences in safety tradeoffs across divisions. In responding to our paper, however, the FDA has said that similar differences in time to approval by drug type are seen at other drug approval agencies. If true, that would be an important criticism.
Fortuitously, some relevant data crossed our desk recently. The Center for Innovation in Regulatory Science (CIRS), a UK based research consortium, compared median review times at the FDA to the next most important drug regulatory agency in the world, the European Medicines Agency (they also look at the Japanese agency). To their credit, the FDA is faster on average than the EMA (thanks PDUFA!). What is relevant for our purposes, however, is to compare differences across divisions.
The CIRS breaks drugs into broader classes than we used but the story they tell for the FDA is similar to ours; anti-cancer drugs, for example, are approved much more quickly than neurology drugs. The story for the EMA, however, is very different than for the FDA. For the EMA all types of drugs are approved in roughly the same amount of time.
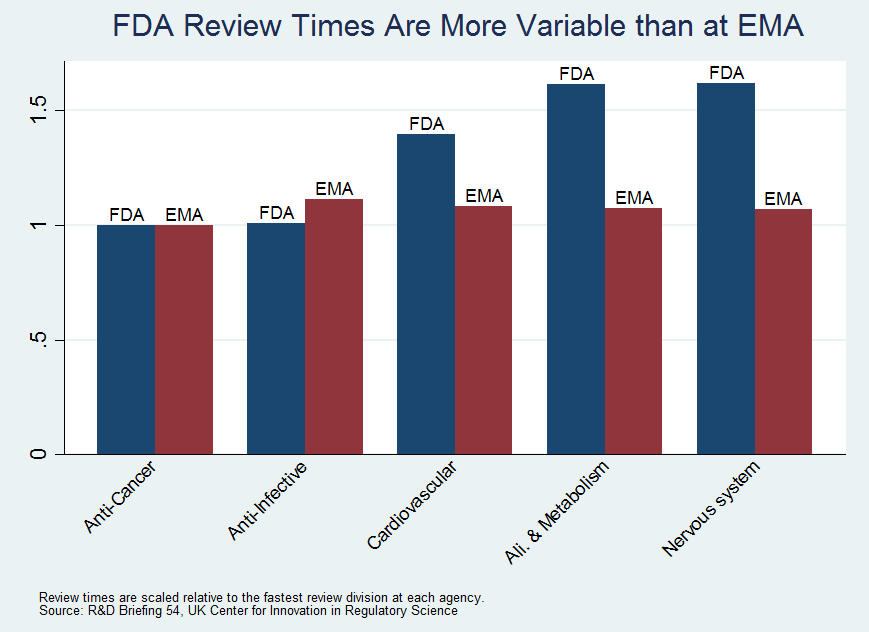
We have argued that the wide variance in performance at the FDA is suggestive of differences in productivity. The fact that we do not see the same wide variance in performance at the EMA is supportive of our argument. Our goal and conclusion still stand:
We support further study to identify the policies and procedures that are working in high-performing divisions, with the goal of finding ways to apply them in low-performing divisions, thereby improving review speed and efficiency.