FDA Device Regulation
In the interests of length I had to sacrifice a few points in my WSJ review of Innovation Breakdown by Joseph Gulfo (excerpted on MR yesterday). In the review, I argued that the FDA could speed the approval of medical devices and reduce uncertainty by not reviewing directly but becoming a certifier of certifiers as is done in Europe.
In fact, a US model is already in place. OSHA, the Occupational Safety and Health, requires that a range of electrical products and materials meet certain safety standards but it outsources certification to Underwriters Laboratories and other Nationally Recognized Testing Laboratories. We could and should do the same for medical devices and for drugs. Indeed, if a device or drug is permitted in a developed, advanced economy such as in Europe, Australia and Japan then I see no reason why it ought not to be provisionally approved in the United States (and vice-versa).
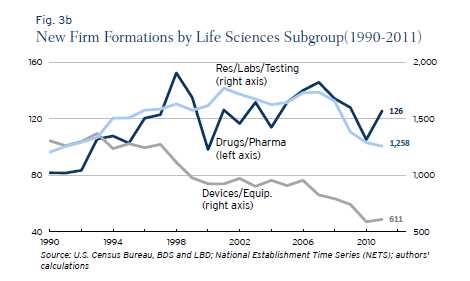
My paper with DiMasi and Milne showed that some FDA drug divisions appear to be much more productive than other divisions suggesting possibilities for substantial improvements if best practices were uniformly adopted. There also appear to be substantial differences between the regulation of drugs and devices especially in recent years. Ian Hathaway and Robert Litan have a new paper on Entrepreneurship and Job Creation in the U.S. Life Sciences Sector that shows that new firm creation in the medical device sector has fallen drastically since 1990 and far more than in the drug sector. Although there are likely many causes, the drop in the number of new firms is consistent with Gulfo’s experience of regulatory uncertainty and may suggest increases in regulatory cost for devices relative to drugs. Here is Hathaway and Litan:
The medical devices and equipment sector, on the other hand, saw new firm formations decline steadily and persistently between 1990 and 2011—falling by 695 firms or 53 percent during that period. Its share of new life sciences firms fell to 31 percent in 2011 from 50 percent in 1990. Unlike its life sciences sector counterparts, the decline in new firm formations in this segment appears to stretch beyond the cyclical effects of the Great Recession.