FDA Deregulation Increases Safety and Innovation and Reduces Prices
In an important and impressive new paper, Parker Rogers looks at what happens when the FDA deregulates or “down-classifies” a medical device type from a more stringent to a less stringent category. He finds that deregulated device types show increases in entry, innovation, as measured by patents and patent quality, and decreases in prices. Safety is either negligibly affected or, in the case of products that come under potential litigation, increased.
After moving from Class III (high regulation) to II (moderate), device types exhibited a 200% increase in patenting and FDA submission rates relative to control groups. Patents filed after these events were also of significantly higher quality, as measured by a 200% increase in received citations and market valuations. These effects do not spill over into similar device types.1 For Class II to I deregulations, the rate of patent filings increased by 50%, though insignificantly, and the quality of patent filings exhibited a significant 10-fold improvement, suggesting that litigation better promotes innovation.
…Down-classification yields considerable benefits, as the proponents of deregulation would predict, but what of product safety? Perhaps counterintuitively, I find that deregulation can improve product safety by exposing firms to more litigation. Despite some adverse event rates increasing after Class III to II events (albeit insignificantly), Class II to I events are associated with significantly lower adverse event rates.3 My analysis of patent texts also reveals that inventors focus more on product safety after deregulation. These results suggest that litigation encourages product safety more than regulation…
Some background. Medical devices are regulated under three categories. New types of devices (new, not necessarily high risk) are highly regulated Class III devices which must go through a pre-market approval process to prove safety and efficacy (like new drugs). The pre-market approval process is time-consuming and expensive but it comes with one significant benefit, federal preemption of state tort action, i.e. these devices are shielded from product liability. Class II devices are devices that are judged to be substantially equivalent to an already approved device–proving equivalence also takes time and money but it’s less onerous than proving safety and efficacy de novo. Note that device manufacturers often make their devices less innovative so they can be approved as Class II devices rather than as Class III devices. Class II devices are mostly also protected against tort litigation. Class I devices are not FDA-approved and are subject to tort litigation.
As experience develops with new devices such that new devices turn out to be not especially risky, the FDA sometimes deregulates or down-classifies these devices from Class III to Class II or from Class II to Class I. Rogers studies these down-classifications by comparing what happens to the down-classified device category to a control group of similar devices that were not down-classified. The control group is critical and Rogers shows that his results are robust to defining the control group in a variety of plausible ways. Some of the key results are shown in the following figure:
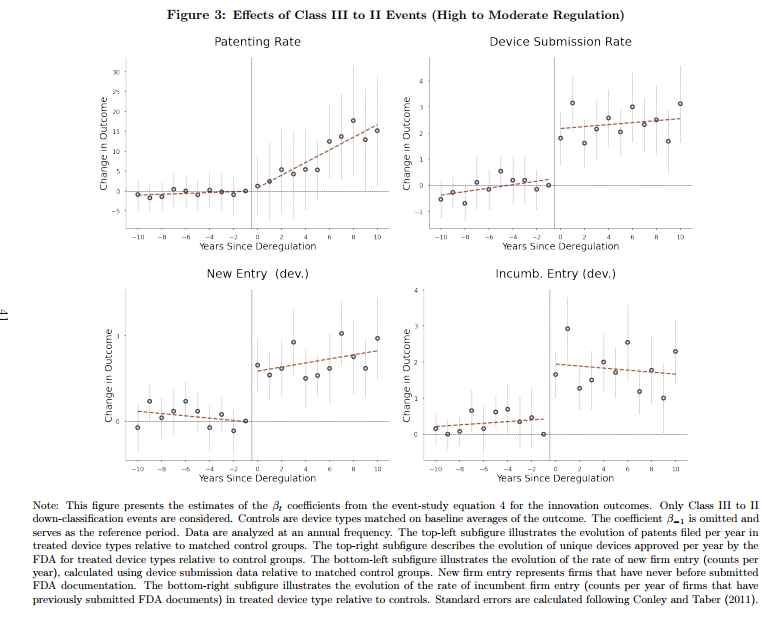 Nicely we see that device submissions and new entry occur very quickly once a device is down-regulated which indicates that firms have ideas and products on-the-shelf but they are dissuaded from entering the market by the onerous pre-market approval process. Most likely, these are products and firms which produce devices for the European market which tends to be less regulated and they enter the US market only when costs are reduced. Patenting also increases in the down-regulated device category and–exactly as one would expect–this takes more time.
Nicely we see that device submissions and new entry occur very quickly once a device is down-regulated which indicates that firms have ideas and products on-the-shelf but they are dissuaded from entering the market by the onerous pre-market approval process. Most likely, these are products and firms which produce devices for the European market which tends to be less regulated and they enter the US market only when costs are reduced. Patenting also increases in the down-regulated device category and–exactly as one would expect–this takes more time.
Safety declines non-significantly if at all from Class III to Class II deregulations and increases for Class II to Class I deregulations. That makes the welfare comparisons easy because deregulation appears to be all benefit and no cost. Note, however, that I have always argued that drugs and devices are actually too safe–that, is we could save more lives on net by approving more drugs and devices even if safety went down. That’s a hard sell, however, but it’s clearly true that given the results here we should deregulate or down-classify many more products even if safety declined on the margin. Too much safety is risky. That’s also the upshot of my paper on off-label prescribing which shows that it’s often the FDA-unapproved off-label use which is the gold-standard treatment in fast moving fields of medicine.
Rogers argues that safety increases for Class II to Class I deregulations because liability is a stronger deterrent on the margin than regulation (and he provides some evidence for this view in that safety increases more among larger firms that are less judgment proof than small firms). Without denying that mechanism my view is that innovation itself increases safety. As I noted above, medical device manufactures often do not use the latest technology in their products because this would threaten the “substantial equivalence” test so you get devices that are actually less safe and also more costly to manufacture than necessary. In essence, substantial equivalence anchors new technologies to old technologies thus preventing movement, even movement towards safety and lower prices.
Rogers also has an excellent and unusual paper (with Jeffrey Clemens) on directed innovation in artificial limbs due to the civil war! That paper and this one show a real focus on digging deep into the data to unearth important and unusual sources of insight. N.B.! Parker Rogers is on the job market.
Addendum: See my many previous posts for more useful references on the FDA, especially Is the FDA Too Conservative or Too Aggressive.