Category: Medicine
A Blueprint for FDA Reform
The new FDA report from Joe Lonsdale and team is impressive. It has a lot of new material, is rich in specifics and bold in vision. Here are just a few of the recommendation which caught my eye:
From the prosaic: GMP is not necessary if you are not manufacturing:
In the U.S., anyone running a clinical trial must manufacture their product under full Good Manufacturing Practices (GMP) regardless of stage. This adds enormous cost (often $10M+) and more importantly, as much as a year’s delay to early-stage research. Beyond the cost and time, these requirements are outright irrational: for example, the FDA often requires three months of stability testing for a drug patients will receive after two weeks. Why do we care if it’s stable after we’ve already administered it? Or take AAV manufacturing—the FDA requires both a potency assay and an infectivity assay, even though potency necessarily reflects infectivity.
This change would not be unprecedented either. By contrast, countries like Australia and China permit Phase 1 trials with non-GMP drug with no evidence of increased patient harm.
The FDA carved out a limited exemption to this requirement in 2008, but its hands are tied by statute from taking further steps. Congress must act to fully exempt Phase 1 trials from statutory GMP. GMP has its place in commercial-scale production. But patients with six months to live shouldn’t be denied access to a potentially lifesaving therapy because it wasn’t made in a facility that meets commercial packaging standards.
Design data flows for AIs:
With modern AI and digital infrastructure, trials should be designed for machine-readable outputs that flow directly to FDA systems, allowing regulators to review data as it accumulates without breaking blinding. No more waiting nine months for report writing or twelve months for post-trial review. The FDA should create standard data formats (akin to GAAP in finance) and waive documentation requirements for data it already ingests. In parallel, the agency should partner with a top AI company to train an LLM on historical submissions, triaging reviewer workload so human attention is focused only where the model flags concern. The goal is simple: get to “yes” or “no” within weeks, not years.
Publish all results:
Clinical trials for drugs that are negative are frequently left unpublished. This is a problem because it slows progress and wastes resources. When negative results aren’t published, companies duplicate failed efforts, investors misallocate capital, and scientists miss opportunities to refine hypotheses. Publishing all trial outcomes — positive or negative—creates a shared base of knowledge that makes drug development faster, cheaper, and more rational. Silence benefits no one except underperforming sponsors; transparency accelerates innovation.
The FDA already has the authority to do so under section 801 of the FDAAA, but failed to adopt a more expansive rule in the past when it created clinicaltrials.gov. Every trial on clincaltrials.gov should have a publication associated with it that is accessible to the public, to benefit from the sacrifices inherent in a patient participating in a clinical trial.
To the visionary:
We need multiple competing approval frameworks within HHS and/or FDA. Agencies like the VA, Medicare, Medicaid, or the Indian Health Service should be empowered to greenlight therapies for their unique populations. Just as the DoD uses elite Special Operations teams to pioneer new capabilities, HHS should create high-agency “SWAT teams” that experiment with novel approval models, monitor outcomes in real time using consumer tech like wearables and remote diagnostics, and publish findings transparently. Let the best frameworks rise through internal competition—not by decree, but by results.
…Clinical trials like the RECOVERY trial and manufacturing efforts like Operation Warp Speed were what actually moved the needle during COVID. That’s what must be institutionalized. Similarly, we need to pay manufacturers to compete in rapidly scaling new facilities for drugs already in shortage today. This capacity can then be flexibly retooled during a crisis.
Right now, there’s zero incentive to rapidly build new drug or device manufacturing plants because FDA reviews move far too slowly. Yet, when crisis strikes, America must pivot instantly—scaling production to hundreds of millions of doses or thousands of devices within weeks, not months or years. To build this capability at home, the Administration and FDA should launch competitive programs that reward manufacturers for rapidly scaling flexible factories—similar to the competitive, market-driven strategies pioneered in defense by the DIU. Speed, flexibility, and scale should be the benchmarks for success, not bureaucratic checklists. While the drugs selected for these competitive efforts shouldn’t be hypothetical—focus on medicines facing shortages right now. This ensures every dollar invested delivers immediate value, eliminating waste and strengthening our readiness for future crises.
To prepare for the next emergency, we need to practice now. That means running fast, focused clinical trials on today’s pressing questions—like the use of GLP-1s in non-obese patients—not just to generate insight, but to build the infrastructure and muscle memory for speed.
Read the whole thing.
Hat tip: Carl Close.
My Conversation with the excellent Sheilagh Ogilvie
Here is the audio, video, and transcript. Here is part of the episode summary:
Tyler and Sheilagh discuss the economic impacts of historical pandemics, the “happy story” of the Black Death and why it doesn’t stand up to scrutiny, the history of variolation and how entrepreneurs created vaccination franchises in 18th-century England, why local communities typically managed epidemics better than central authorities, the dastardly nature of medieval guilds, the European marriage pattern and its disputed contribution to economic growth, when sustained economic growth truly began in England, why the Dutch Republic stagnated despite its early success, whether she agrees with Greg Clark’s social mobility hypothesis, her experience and conducting “anthropological fieldwork” on English social customs, the communitarian norms she encountered while living in Germany, her upcoming research project on European serfdom, and more.
Here is one excerpt:
OGILVIE: …If you were a teenager in an English village in the 18th century and you were deciding, “I’m going to move to London and get a job,” you and your friendship group from the village would all go into the nearest town and pay a commercial variolator. You’d all get smallpox together. You’d go back to your village. You’d suffer through this mild case of smallpox, and then you would be immunized for life, assuming that you hadn’t died. You would go off to London and seek your fortune. It was very much a normal teenage thing to do.
There was this incredible franchising set up in England. It was like a McDonald’s, but to get variolated. There were these entrepreneurs who advertised themselves as having lower-risk ways of getting immunized and cheaper ways of getting immunized. There was this famous family of the Suttons that started a franchise in 18th-century England in the 1750s. Then they spread into the continent of Europe and actually into North America.
COWEN: You would have done it back then?
OGILVIE: Oh, definitely.
COWEN: With enthusiasm.
And this:
COWEN: You’ve now lived in England for well over 30 years. What’s been your biggest surprise about the place, if anything has stuck?
OGILVIE: It keeps on surprising me. I’ve actually lived here for more than 46 years. I moved here as an undergraduate. I came here when I was 16, and I feel as if I’m still doing anthropological fieldwork on the behavioral patterns of these strange local tribes. There are these systematic things — they’re charming, but they’re very strange.
For instance, just to give one example, English people are very reserved. I get on with that because Canadians are fairly reserved as well. It’s okay to talk to people in your neighborhood if they have a dog with them. That’s a conversation mediator. Or if you are gardening in your front garden, but if you’re in your back garden, you’re not supposed to talk to people. It’s taken me a few decades to observe this as an empirical regularity.
Nobody ever tells you that this is how you’re supposed to behave, but if you keep your field notebooks going as an anthropologist, you begin to notice the tribal patterns of the English. I must like them, since here I still am after more than four decades.
Recommended.
Federal Judge Rejects FDA Power Grab
In Don’t Let the FDA Regulate Lab Tests! and The New FDA and the Regulation of Laboratory Developed Tests I warned that the FDA’s power grab over laboratory developed tests was both unlawful and likely to result in deadly harm (as it did during COVID). Thus, I am pleased that a Federal judge has vacated the FDA’s rule entirely, writing:
…the text, structure, and history of the FDCA and CLIA make clear that FDA lacks the authority to regulate laboratory-developed test services.
…FDA’s asserted jurisdiction over laboratory-developed test services as “devices” under the FDCA defies bedrock principles of statutory interpretation, common sense, and longstanding industry practice.
The judge also noted some of the costs that I had pointed to:
…the Fifth Circuit has made clear that district courts should generally “nullify and revoke” illegal agency action, Braidwood, 104 F.4th at 951. The Court finds that such relief is appropriate here. The final rule will initially impact nearly 80,000 existing tests offered by almost 1,200 laboratories, and it will also affect about 10,013 new tests offered every year going forward. The estimated compliance costs for laboratories across the country will total well over $1 billion per year, and over the next two decades, FDA projects that total costs associated with the rule will range from $12.57 billion to $78.99 billion. FDA acknowledges that the enormous increased costs to laboratories may cause price increases and reduce the amount of revenue a laboratory can invest in creating and modifying tests.
… For these reasons, it is ORDERED that the Laboratory Plaintiffs’ Motions for Summary Judgment, (Dkt. #20, #27), are GRANTED. The final rule is hereby SET ASIDE and VACATED.
HHS head RFK Jr. should immediately instruct the FDA to halt any further efforts to regulate laboratory developed tests.
AI Discovers New Uses for Old Drugs
The NYTimes has an excellent piece by Kate Morgan on AI discovering new uses for old drugs:
A little over a year ago, Joseph Coates was told there was only one thing left to decide. Did he want to die at home, or in the hospital?
Coates, then 37 and living in Renton, Wash., was barely conscious. For months, he had been battling a rare blood disorder called POEMS syndrome, which had left him with numb hands and feet, an enlarged heart and failing kidneys. Every few days, doctors needed to drain liters of fluid from his abdomen. He became too sick to receive a stem cell transplant — one of the only treatments that could have put him into remission.
“I gave up,” he said. “I just thought the end was inevitable.”
But Coates’s girlfriend, Tara Theobald, wasn’t ready to quit. So she sent an email begging for help to a doctor in Philadelphia named David Fajgenbaum, whom the couple met a year earlier at a rare disease summit.
By the next morning, Dr. Fajgenbaum had replied, suggesting an unconventional combination of chemotherapy, immunotherapy and steroids previously untested as a treatment for Coates’s disorder.
Within a week, Coates was responding to treatment. In four months, he was healthy enough for a stem cell transplant. Today, he’s in remission.
The lifesaving drug regimen wasn’t thought up by the doctor, or any person. It had been spit out by an artificial intelligence model.
AI is excellent at combing through large amounts of data to find surprising connections.
Discovering new uses for old drugs has some big advantages and one disadvantage. A big advantage is that once a drug has been approved for some use it can be prescribed for any use–thus new uses of old drugs do not have to go through the lengthy and arduous FDA approval procedures. In essence, off-label uses have been safety-tested but not FDA efficacy-tested in the new use. I use this fact about off-label prescribing to evaluate the FDA. During COVID, for example, the British Recovery trial, discovered that the common drug, dexamethasone could reduce mortality by up to one-third in hospitalized patients on oxygen support that knowledge was immediately applied, saving millions of lives worldwide:
Within hours, the result was breaking news across the world and hospitals were adopting the drug into the standard care given to all patients with COVID-19. In the nine months following the discovery, dexamethasone saved an estimated one million lives worldwide.
New uses for old drugs are typically unpatentable, which helps keep them cheap—but the disadvantage is that this also weakens private incentives to discover them. While FDA trials for these new uses are often unnecessary, making development costs much lower, the lack of strong market protection can still deter investment. The FDA offers some limited exclusivity through programs like 505(b)(2), which grants temporary protection for new clinical trials or safety and efficacy data. These programs are hard to calibrate—balancing cost and reward is difficult—but likely provide some net benefits.
The NIH should continue prioritizing research into unpatentable treatments, as this is where the market is most challenged. More broadly, research on novel mechanisms to support non-patentable innovations is valuable. That said, I’m not overly concerned about under-investment in repurposing old drugs, especially as AI further reduces the cost of discovery.
Peter Marks Forced Out at FDA
Peter Marks was key to President Trump’s greatest first-term achievement: Operation Warp Speed. In an emergency, he pushed the FDA to move faster—against every cultural and institutional incentive to go slow. He fought the system and won.
I had some hope that FDA commissioner Marty Makary would team with Marks at CBER. Makary understands that the FDA moves too slowly. He wrote in 2021:
COVID has given us a clear-eyed look at a broken Food and Drug Administration that’s mired in politics and red tape.
Americans can now see why medical advances often move at turtle speed. We need fresh leadership at the FDA to change the culture at the agency and promote scientific advancement, not hinder it.
This starts at the top. Our public health leaders have become too be accepting of the bureaucratic processes that would outrage a fresh eye. For example, last week the antiviral pill Molnupiravir was found to cut COVID hospitalizations in half and, remarkably, no one who got the drug died.
The irony is that Molnupiravir was developed a year ago. Do the math on the number of lives that could have been saved if health officials would have moved fast, allowing rolling trials with an evaluation of each infection and adverse event in real-time. Instead, we have a process that resembles a 7-part college application for each of the phase 1, 2, and 3 clinical trials.
A Makary-Marks team could have moved the FDA in a very promising direction. Unfortunately, disputes with RFK Jr proved too much. Marks was especially and deservedly outraged by the measles outbreak and the attempt to promote vitamins over vaccines:
“It has become clear that truth and transparency are not desired by the Secretary, but rather he wishes subservient confirmation of his misinformation and lies,” Marks wrote in a resignation letter referring to HHS Secretary Robert F. Kennedy Jr.
Thus, as of now, the FDA is moving in the wrong direction and Makary has lost an ally against RFK.
In other news, the firing of FDA staff is slowing down approvals, as I predicted it would.
What Follows from Lab Leak?
Does it matter whether SARS-CoV-2 leaked from a lab in Wuhan or had natural zoonotic origins? I think on the margin it does matter.
First, and most importantly, the higher the probability that SARS-CoV-2 leaked from a lab the higher the probability we should expect another pandemic.* Research at Wuhan was not especially unusual or high-tech. Modifying viruses such as coronaviruses (e.g., inserting spike proteins, adapting receptor-binding domains) is common practice in virology research and gain-of-function experiments with viruses have been widely conducted. Thus, manufacturing a virus capable of killing ~20 million human beings or more is well within the capability of say ~500-1000 labs worldwide. The number of such labs is growing in number and such research is becoming less costly and easier to conduct. Thus, lab-leak means the risks are larger than we thought and increasing.
A higher probability of a pandemic raises the value of many ideas that I and others have discussed such as worldwide wastewater surveillance, developing vaccine libraries and keeping vaccine production lines warm so that we could be ready to go with a new vaccine within 100 days. I want to focus, however, on what new ideas are suggested by lab-leak. Among these are the following.
Given the risks, a “Biological IAEA” with similar authority as the International Atomic Energy Agency to conduct unannounced inspections at high-containment labs does not seem outlandish. (Indeed the Bulletin of Atomic Scientists are about the only people to have begun to study the issue of pandemic lab risk.) Under the Biological Weapons Convention such authority already exists but it has never been used for inspections–mostly because of opposition by the United States–and because the meaning of biological weapon is unclear, as pretty much everything can be considered dual use. Notice, however, that nuclear weapons have killed ~200,000 people while accidental lab leak has probably killed tens of millions of people. (And COVID is not the only example of deadly lab leak.) Thus, we should consider revising the Biological Weapons Convention to something like a Biological Dangers Convention.
BSL3 and especially BSL4 safety procedures are very rigorous, thus the issue is not primarily that we need more regulation of these labs but rather to make sure that high-risk research isn’t conducted under weaker conditions. Gain of function research of viruses with pandemic potential (e.g. those with potential aerosol transmissibility) should be considered high-risk and only conducted when it passes a review and is done under BSL3 or BSL4 conditions. Making this credible may not be that difficult because most scientists want to publish. Thus, journals should require documentation of biosafety practices as part of manuscript submission and no journal should publish research that was done under inappropriate conditions. A coordinated approach among major journals (e.g., Nature, Science, Cell, Lancet) and funders (e.g. NIH, Wellcome Trust) can make this credible.
I’m more regulation-averse than most, and tradeoffs exist, but COVID-19’s global economic cost—estimated in the tens of trillions—so vastly outweighs the comparatively minor cost of upgrading global BSL-2 labs and improving monitoring that there is clear room for making everyone safer without compromising research. Incredibly, five years after the crisis and there has be no change in biosafety regulation, none. That seems crazy.
Many people convinced of lab leak instinctively gravitate toward blame and reparations, which is understandable but not necessarily productive. Blame provokes defensiveness, leading individuals and institutions to obscure evidence and reject accountability. Anesthesiologists and physicians have leaned towards a less-punitive, systems-oriented approach. Instead of assigning blame, they focus in Morbidity and Mortality Conferences on openly analyzing mistakes, sharing knowledge, and redesigning procedures to prevent future harm. This method encourages candid reporting and learning. At its best a systems approach transforms mistakes into opportunities for widespread improvement.
If we can move research up from BSL2 to BSL3 and BSL4 labs we can also do relatively simple things to decrease the risks coming from those labs. For example, let’s not put BSL4 labs in major population centers or in the middle of a hurricane prone regions. We can also, for example, investigate which biosafety procedures are most effective and increase research into safer alternatives—such as surrogate or simulation systems—to reduce reliance on replication-competent pathogens.
The good news is that improving biosafety is highly tractable. The number of labs, researchers, and institutions involved is relatively small, making targeted reforms feasible. Both the United States and China were deeply involved in research at the Wuhan Institute of Virology, suggesting at least the possibility of cooperation—however remote it may seem right now.
Shared risk could be the basis for shared responsibility.
Bayesian addendum *: A higher probability of a lab-leak should also reduce the probability of zoonotic origin but the latter is an already known risk and COVID doesn’t add much to our prior while the former is new and so the net probability is positive. In other words, the discovery of a relatively new source of risk increases our estimate of total risk.
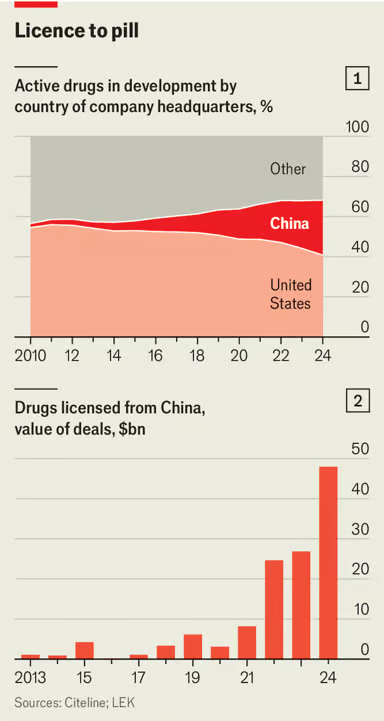
China’s Medicines are Saving American Lives
The Economist reports that China is now the second largest producer of new pharmaceuticals, after the United States.
China has long been known for churning out generic drugs, supplying raw ingredients and managing clinical trials for the pharmaceutical world. But its drugmakers are now also at the cutting edge, producing innovative medicines that are cheaper than the ones they compete with.
… In September last year an experimental drug did what none had done before. In late-stage trials for non-small cell lung cancer, it nearly doubled the time patients lived without the disease getting worse—to 11.1 months, compared with 5.8 months for Keytruda. The results were stunning. So too was the nationality of the biotech company behind them. Akeso is Chinese.
This is exactly what I predicted in my TED talk and it’s great news! As I said then:
Ideas have this amazing property. Thomas Jefferson said “He who receives an idea from me receives instruction himself, without lessening mine. As he who lights his candle at mine receives light without darkening me.”
Now think about the following: if China and India were as rich as the United States is today, the market for cancer drugs would be eight times larger than it is now. Now we are not there yet, but it is happening. As other countries become richer the demand for these pharmaceuticals is going to increase tremendously. And that means an increase incentive to do research and development, which benefits everyone in the world. Larger markets increase the incentive to produce all kinds of ideas, whether it’s software, whether it’s a computer chip, whether it’s a new design.
Well if larger markets increase the incentive to produce new ideas, how do we maximize that incentive?
It’s by having one world market, by globalizing the world. Ideas are meant to be shared.
One idea, one world, one market.
Sadly, some of us are losing sight of the immense benefits of a global market. Another example of the great forgetting.
As Girard predicted, China’s growing similarity to the U.S. has fueled conflict and rivalry. But if managed properly, rivalry can be positive-sum. A rich China benefits us far more than a poor China—including by creating new cancer medicines that save American lives.
Hat tip: Cremieux.
My excellent Conversation with Ezra Klein
Ezra is getting plenty of coverage for his very good and very on the mark new book with Derek Thompson, Abundance. So far it is a huge hit after only a few days. I figured this conversation would be most interesting, and add the most value, if I tried to push him further from a libertarian point of view (a sign of respect of course). Here is the audio, video, and transcript. Here is part of the episode summary:
In this conversation, Ezra and Tyler discuss how the abundance agenda interacts with political polarization, whether it’s is an elite-driven movement, where Ezra favors NIMBYism, the geographic distribution of US cities, an abundance-driven approach to health care, what to do about fertility decline, how the U.S. federal government might prepare for AGI, whether mass layoffs in government are justified, Ezra’s recommended travel destinations, and more.
Lots of good back and forth, here is one excerpt:
COWEN: Here’s a question from a reader, and I’m paraphrasing. “I can see why you would favor Obamacare and an abundance agenda because Obamacare throws a lot more resources at the healthcare sector in some ways. It did have Medicare cuts, but nonetheless, it’s not choking the sector. But if you favor an abundance agenda, can you then possibly favor single-payer health insurance through the government, which does tend to choke resources and stifle innovation?”
KLEIN: I think it would depend on how you did the single-payer healthcare. Here, we should talk about — because it’s referenced glancingly in the book in a place where you and I differ — but the supervillain view that I hold and your view, which is that you should negotiate drug prices. I’ve always thought on that because I think in some ways, it’s a better toy example than single payer versus Obamacare.
I think you want to take the amount of innovation you’re getting very, very, very seriously. I’ve written pieces about this, that I think if you’re going to do Medicare drug pricing at any kind of significant level, you want to be pairing that with a pretty significant agenda to make drug discovery much easier, to make testing much easier.
And:
COWEN: What should the US federal government do to prepare for AGI? We should just lay off people, right?
KLEIN: [laughs] I would not say it that way. I wouldn’t say just lay off people. I think that’s some of what we’re doing.
COWEN: No, not just, but step one.
KLEIN: Do you think that’s step one? Do you buy this DOGE’s preparation-for-AGI argument that you hear?
COWEN: I think maybe a fifth of them think that. Maybe it’s step two or step three, but it’s a pretty early step, right?
KLEIN: I think that the question of AI or AGI in the federal government, in anywhere — and this is one reason I’ve not bought this argument about DOGE — is you have to ask, “Well what is this AI or AGI doing? What is its value function? What prompt have you given it? What have you asked it to execute across the government and how?”
Alignment, which we have primarily talked about in terms of whether or not the AI, the superintelligence makes us all into paperclips, is a constant question of just near-term systems as well. I think the question of how should we prepare for AGI or for AI in the federal government first has to do with deciding what we would like the AI or the AGI to do. That could be different things to different areas.
My sense — talking to a bunch of people in the companies has helped me conceptualize this better — is that the first thing I would do is begin to ask, what do I think the opportunities of AI are, scientifically and in terms of different kinds of discoveries…
And this:
COWEN: Let me give you another right-wing view, and tell me what you think. The notion that the most important feature of state capacity is whether a state has enough of its citizens willing to fight and die for it. In that case, the United States, Israel, but a pretty small number of nations have high state capacity, and most of Western Europe really does not because they don’t have militaries that mean anything. Is that just the number one feature of abundance in state capacity?
Recommended, obviously.
What Did We Learn From Torturing Babies?
As late as the 1980s it was widely believed that babies do not feel pain. You might think that this was an absurd thing to believe given that babies cry and exhibit all the features of pain and pain avoidance. Yet, for much of the 19th and 20th centuries, the straightforward sensory evidence was dismissed as “pre-scientific” by the medical and scientific establishment. Babies were thought to be lower-evolved beings whose brains were not yet developed enough to feel pain, at least not in the way that older children and adults feel pain. Crying and pain avoidance were dismissed as simply reflexive. Indeed, babies were thought to be more like animals than reasoning beings and Descartes had told us that an animal’s cries were of no more import than the grinding of gears in a mechanical automata. There was very little evidence for this theory beyond some gesturing’s towards myelin sheathing. But anyone who doubted the theory was told that there was “no evidence” that babies feel pain (the conflation of no evidence with evidence of no effect).
Most disturbingly, the theory that babies don’t feel pain wasn’t just an error of science or philosophy—it shaped medical practice. It was routine for babies undergoing medical procedures to be medically paralyzed but not anesthetized. In one now infamous 1985 case an open heart operation was performed on a baby without any anesthesia (n.b. the link is hard reading). Parents were shocked when they discovered that this was standard practice. Publicity from the case and a key review paper in 1987 led the American Academy of Pediatrics to declare it unethical to operate on newborns without anesthesia.
In short, we tortured babies under the theory that they were not conscious of pain. What can we learn from this? One lesson is humility about consciousness. Consciousness and the capacity to suffer can exist in forms once assumed to be insensate. When assessing the consciousness of a newborn, an animal, or an intelligent machine, we should weigh observable and circumstantial evidence and not just abstract theory. If we must err, let us err on the side of compassion.
Claims that X cannot feel or think because Y should be met with skepticism—especially when X is screaming and telling you different. Theory may convince you that animals or AIs are not conscious but do you want to torture more babies? Be humble.
We should be especially humble when the beings in question are very different from ourselves. If we can be wrong about animals, if we can be wrong about other people, if we can be wrong about our own babies then we can be very wrong about AIs. The burden of proof should not fall on the suffering being to prove its pain; rather, the onus is on us to justify why we would ever withhold compassion.
Hat tip: Jim Ward for discussion.
The Shortage that Increased Ozempic Supply
It sometimes happens that a patient needs a non-commercially-available form of a drug, a different dosage or a specific ingredient added or removed depending on the patient’s needs. Compounding pharmacies are allowed to produce these drugs without FDA approval. Moreover, since the production is small-scale and bespoke the compounded drugs are basically immune from any patent infringement claims. The FDA, however, also has an oddly sensible rule that says when a drug is in shortage they will allow it be compounded, even when the compounded version is identical to the commercial version.
The shortage rule was meant to cover rare drugs but when demand for the GLP-1 drugs like Ozempic and Zepbound skyrocketed, the FDA declared a shortage and big compounders jumped into the market offering these drugs at greatly reduced prices. Moreover, the compounders advertised heavily and made it very easy to get a “prescription.” Thus, the GLP-1 compounders radically changed the usual story where the patient asks the compounder to produce a small amount of a bespoke drug. Instead the compounders were selling drugs to millions of patients.
Thus, as a result of the shortage rule, the shortage led to increased supply! The shortage has now ended, however, which means you can expect to see many fewer Hims and Hers ads.
Scott Alexander makes an interesting point in regard to this whole episode:
I think the past two years have been a fun experiment in semi-free-market medicine. I don’t mean the patent violations – it’s no surprise that you can sell drugs cheap if you violate the patent – I mean everything else. For the past three years, ~2 million people have taken complex peptides provided direct-to-consumer by a less-regulated supply chain, with barely a fig leaf of medical oversight, and it went great. There were no more side effects than any other medication. People who wanted to lose weight lost weight. And patients had a more convenient time than if they’d had to wait for the official supply chain to meet demand, get a real doctor, spend thousands of dollars on doctors’ visits, apply for insurance coverage, and go to a pharmacy every few weeks to pick up their next prescription. Now pharma companies have noticed and are working on patent-compliant versions of the same idea. Hopefully there will be more creative business models like this one in the future.
The GLP-1 drugs are complex peptides and the compounding pharmacies weren’t perfect. Nevertheless, I agree with Scott that, as with the off-label market, the experiment in relaxed FDA regulation was impressive and it does provide a window onto what a world with less FDA regulation would look like.
Hat tip: Jonathan Meer.
More British DOGE
Sir Keir Starmer is abolishing NHS England as Labour embarks on the biggest reorganisation of the health service for more than a decade.
The prime minister said that scrapping the arm’s-length body would bring “management of the NHS back into democratic control” and reduce spending on “two layers of bureaucracy”.
He said the quango, responsible for the day-to-day running of the health service, was the ultimate example of “politicians almost not trusting themselves, outsourcing everything to different bodies … to the point you can’t get things done”.
Starmer argued: “I don’t see why the decision about £200 billion of taxpayer money on something as fundamental to our security as the NHS should be taken by an arm’s-length body.”
NHS England will now be brought back under the control of the Department of Health and Social Care (DHSC), and the two organisations will be merged over the next two years, leading to about 10,000 job cuts.
Here is more from the Times of London.
Germany fact of the day
Germany opened its doors a decade ago to nearly 1 million Syrians, taking in more than any other country in Europe. Today, some 6,000 Syrian doctors make up the single largest group of foreign-born physicians, filling vital gaps in care at hospitals and clinics from the Alps to the Baltic Sea. That is especially true in rural areas, where attracting doctors can be hard. But even in big cities, Syrian doctors now make up the majority of attending physicians at some medical practices.
Here is more from The Washington Post. Here is my previous post on Syrians in Germany.
Visits to the Doctor, Per Year
The number of times people visit the doctor per year varies tremendously across OECD countries from a low of 2.9 in Chile to a high of 17.5 (!) in Korea. I haven’t run the numbers officially but it doesn’t seem that there is much correlation with medical spending per capita or life expectancy.
Data can be found here. 
Hat tip: Emil Kirkegaard on X.
My Conversation with Carl Zimmer
Here is the audio, video, and transcript. Here is part of the episode summary:
He joins Tyler to discuss why it took scientists so long to accept airborne disease transmission and more, including why 19th-century doctors thought hay fever was a neurosis, why it took so long for the WHO and CDC to acknowledge COVID-19 was airborne, whether ultraviolet lamps can save us from the next pandemic, how effective masking is, the best theory on the anthrax mailings, how the U.S. military stunted aerobiology, the chance of extraterrestrial life in our solar system, what Lee Cronin’s “assembly theory” could mean for defining life itself, the use of genetic information to inform decision-making, the strangeness of the Flynn effect, what Carl learned about politics from growing up as the son of a New Jersey congressman, and much more.
Here is an excerpt:
COWEN: Over time, how much will DNA information enter our daily lives? To give a strange example, imagine that, for a college application, you have to upload some of your DNA. Now to unimaginative people, that will sound impossible, but if you think about the equilibrium rolling itself out slowly — well, at first, students disclose their DNA, and over time, the DNA becomes used for job hiring, for marriage, in many other ways. Is this our future equilibrium, that genetic information will play this very large role, given how many qualities seem to be at least 40 percent to 60 percent inheritable, maybe more?
ZIMMER: The term that a scientist in this field would use would be heritable, not inheritable. Inheritability is a slippery thing to think about. I write a lot about that in my book, She Has Her Mother’s Laugh, which is about heredity in general. Heritability really is just saying, “Okay, in a certain situation, if I look at different people or different animals or different plants, how much of their variation can I connect with variation in their genome?” That’s it. Can you then use that variability to make predictions about what’s going to happen in the future? That is a totally different question in many —
COWEN: But it’s not totally different. Your whole family’s super smart. If I knew nothing about you, and I knew about the rest of your family, I’d be more inclined to let you into Yale, and that would’ve been a good decision. Again, only on average, but just basic statistics implies that.
ZIMMER: You’re very kind, but what do you mean by intelligent? I’d like to think I’m pretty good with words and that I can understand scientific concepts. I remember in college getting to a certain point with calculus and being like, “I’m done,” and then watching other people sail on.
COWEN: Look, you’re clearly very smart. The New York Times recognizes this. We all know statistics is valid. There aren’t any certainties. It sounds like you’re running away from the science. Just endorse the fact you came from a very smart family, and that means it’s quite a bit more likely that you’ll be very smart too. Eventually, the world will start using that information, would be the auxiliary hypothesis. I’m asking you, how much will it?
ZIMMER: The question that we started with was about actually uploading DNA. Then the question becomes, how much of that information about the future can you get out of DNA? I think that you just have to be incredibly cautious about jumping to conclusions about it because the genome is a wild and woolly place in there, and the genome exists in environments. Even if you see broad correlations on a population level, as a college admission person, I would certainly not feel confident just scanning someone’s DNA for information in that regard.
COWEN: Oh, that wouldn’t be all you would do, right? They do plenty of other things now. Over time, say for job hiring, we’ll have the AI evaluate your interview, the AI evaluate your DNA. It’ll be highly imperfect, but at some point, institutions will start doing it, if not in this country, somewhere else — China, Singapore, UAE, wherever. They’re not going to be so shy, right?
ZIMMER: I can certainly imagine people wanting to do that stuff regardless of the strength of the approach. Certainly, even in the early 1900s, we saw people more than willing to use ideas about inherited levels of intelligence to, for example, decide which people should be institutionalized, who should be allowed into the United States or not.
For example, Jews were considered largely to be developmentally disabled at one point, especially the Jews from Eastern Europe. We have seen that people are certainly more than eager to jump from the basic findings of DNA to all sorts of conclusions which often serve their own interests. I think we should be on guard that we not do that again.
And:
COWEN: If we take the entirety of science, you’ve written on many topics in a very useful way, science policy. Where do you think your views are furthest from the mainstream or the orthodoxy? Where do you have the weirdest take relative to other people you know and respect? I think we should just do plenty of human challenge trials. That would be an example of something you might say, but what would the answer be for you?
I very much enjoyed Carl’s latest book Air-Borne: The Hidden History of the Air We Breathe.
Hire Don’t Fire at the FDA
As a longtime critic of the FDA, you might expect me to support firing FDA employees—not so! My focus has always been on reducing approval time and costs to speed drugs to patients and increase the number of new drugs. Cutting staff is more likely to slow approvals and raise costs.
To be fair, we’re talking about the firing of some 200 probationary employees from a total of some 20,000. Unusual but not earth shaking. But the firings are indiscriminate, and as I explain below, the FDA is a peculiar target for cost-cutting because user fees under PDUFA cover a significant share of the FDA’s budget so its workers are among the cheapest federal employees. So what is the point? Shock and awe in advance of bigger reforms for the FDA? Perhaps. Regardless, I think we should keep in mind the big picture on staff and speed.
The Prescription Drug User Fee Act of 1992 (PDUFA) provides strong evidence that with more staff the FDA works faster to get new and better drugs to patients. Before PDUFA, drug approvals languished at the FDA simply due to a lack of staff—harming both drug companies and patients. Congress should have increased FDA funding, as the benefits would have far outweighed the costs, but Congress failed. Instead, PDUFA created a workaround: drug firms agreed to pay user fees, with the condition that the funds be used for drug reviewers and that the FDA be held to strict review standards.
PDUFA was a tremendous success. Carpenter et al., Olson, Berndt et al. and others all find that PDUFA shortened review times and it did so primarily through the mechanism of hiring more staff. Thus, Carpenter et al. report “NDA review times shortened by 3.3 months for every 100 additional FDA staff.” Moreover, the faster approval times came at little to no expense of reduced safety. Thus, Berndt et al. report:
implementation of the PDUFAs led to substantial incremental reductions in approval times beyond what would have been observed in the absence of these legislative acts. In addition, our preliminary examination of the trends in the number of new molecular entity withdrawals, frequently used as a proxy to assess the FDA’s safety record, suggests that the proportion of approvals ultimately leading to safety withdrawals prior to PDUFA and during PDUFA I and II were not statistically different.
And in a later analysis Philipson et al. find that:
more rapid access of drugs on the market enabled by PDUFA saved the equivalent of 140,000 to 310,000 life years. Additionally, we estimate an upper bound on the adverse effects of PDUFA based on drugs submitted during PDUFA I/II and subsequently withdrawn for safety reasons, and find that an extreme upper bound of about 56,000 life years were lost. This estimate is an extreme upper bound as it assumes all withdrawals since the inception of PDUFA were due to PDUFA and that there were no patients who benefitted from the withdrawn drugs.
If we’re going to have FDA review, it should be fast and efficient. We need to shift the focus from the FDA’s balance sheet in the Federal budget to the patients it serves—more staff means faster reviews, better access to treatments, and a healthier society.
More generally, government regulation, not staffing, is the real problem. Cut regulation, and staff cuts can follow. Cut staff without cutting regulation, and the morass only gets worse.